| Dermatofibrosarcoma protuberans | |
|---|---|
| Other names | DFSP [1] |
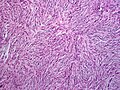
_recurrence.JPG) | |
| Histopathological image of dermatofibrosarcoma protuberans. Local recurrence long after the first excision. H&E stain | |
| Specialty | Oncology |
Dermatofibrosarcoma protuberans (DFSP)[2] is a rare locally aggressive malignant cutaneous soft-tissue sarcoma. DFSP develops in the connective tissue cells in the middle layer of the skin (dermis).[3] Estimates of the overall occurrence of DFSP in the United States are 0.8 to 4.5 cases per million persons per year.[4][5] In the United States, DFSP accounts for between 1 and 6 percent of all soft-tissue sarcomas[6] and 18 percent of all cutaneous soft-tissue sarcomas. In the Surveillance, Epidemiology and End Results (SEER) tumor registry from 1992 through 2004, DFSP was second only to Kaposi sarcoma.
Presentation
[edit]Dermatofibrosarcoma protuberans begins as a minor firm area of skin most commonly about to 1 to 5 cm in diameter. It can resemble a bruise, birthmark, or pimple. It is a slow-growing tumor and is usually found on the torso but can occur anywhere on the body.[7] About 90% of DFSPs are low-grade sarcomas. About 10% are mixed, containing a high-grade sarcomatous component (DFSP-FS); therefore, they are considered to be intermediate-grade sarcomas. DFSPs rarely lead to a metastasis (fewer than 5% metastasize), but DFSPs can recur locally. DFSPs most often arise in patients who are in their thirties but this may be due to diagnostic delay.
Location
[edit]Commonly located on the chest and shoulders, the following is the site distribution of DFPS as was observed in Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2010.[5][8]
- Trunk/torso – 42%
- Lower extremity – 21%
- Upper extremity – 21%
- Head and neck – 13%
- Genitals – 1%
Variants
[edit]The World Health Organization in 2020 classified the fibro sarcomatous DFSP (DFSP-FS) variant (also termed dermatofibrosarcoma protuberans, fibro sarcomatous) of the dermatofibrosarcoma protuberans as a specific form of the intermediate (rarely metastasizing) fibroblastic and myofibroblastic tumors and other variants of this disorder as a specific form of the intermediate (locally aggressive) fibroblastic and myofibroblastic tumors.[9]
Bednar tumors
[edit]Bednar, or pigmented DFSP, is distinguished by the dispersal of melanin-rich dendritic cells of the skin. It represents 1–5 percent[10] of all DFSP occurring in people rich in melanin pigments. Bednar is characterized by a dermal spindle cell proliferation like DFSP but distinguished by the additional presence of melanocytic dendritic cells. It occurs at the same rate as DFSP on fairer skin and should be considered to have the same chances of metastasis.[11]
Myxoid DFSP
[edit]Myxoid DFSP has areas of myxoid degeneration in the stroma.[12][13]
Giant cell fibroblastoma
[edit]Giant cell fibroblastoma[2] contains giant cells, and is also known as juvenile DFSP.[14] Giant cell fibroblastomas are skin and soft-tissue tumors that usually arise in childhood. They are sometimes seen in association with dermatofibrosarcoma protuberans (DFSP, hybrid lesions) or may transform or recur as DFSP.[15][13]
Atrophic DFSP
[edit]Atrophic DFSP resemble other benign lesions such as morphea, idiopathic atrophoderma, atrophic scar, anetoderma or lipoatrophy. It behaves like classic DFSP. It commonly favours young to middle-aged adults. It has a slow infiltrative growth and a high rate of local recurrence if not completely excised.[13][16]
Sclerosing DFSP
[edit]Sclerosing DFSP is a variant in which the cellularity is low, and the tumor consists of uniform bundles of collagen interspersed with more typical DFSP cells.[13]
Granular cell variant is a rare type in which spindle cells are mingled with richly granular cells, the granules being lysosomal, with prominent nucleoli.[13]
Fibrosarcomatous DFSP (DFSP-FS)
[edit]Fibrosarcomatous DFSP is a rare variant of DFSP involving greater aggression, high rates of local occurrences, and higher metastatic potential.[17] DFSP-FS are considered to be intermediate-grade sarcomas,[18] although they rarely metastasize (fewer than 5 percent of cases).
Pathophysiology
[edit]More than 90% of DFSP tumors have the chromosomal translocation t(17;22). The translocation fuses the collagen gene (COL1A1) with the platelet-derived growth factor (PDGF) gene. The fibroblast, the cell of origin of this tumor, expresses the fusion gene in the belief that it codes for collagen. However, the resulting fusion protein is processed into a mature platelet-derived growth factor which is a potent growth factor. Fibroblasts contain the receptor for this growth factor. Thus the cell "thinks" it is producing a structural protein, but it creates a self-stimulatory growth signal. The cell divides rapidly and tumor forms.
The tissue is often positive for CD34.[19][20]
Diagnosis
[edit]
DFSP is a malignant tumor diagnosed with a biopsy, when a portion of the tumor is removed for examination. In order to ensure that enough tissue is removed to make an accurate diagnosis, the initial biopsy of a suspected DFSP is usually done with a core needle or a surgical incision.[21]
Clinical palpation is not entirely reliable for ascertaining the depth of a DFSP infiltration. Magnetic resonance imaging (MRI) is more sensitive addressing the depth of the invasion on some types of DFSP, particularly large or recurring tumors,[22][23] though MRI is less accurate for identifying infiltration to head and neck tumors.
Diagnostic delay and misdiagnosis
[edit]Due to the rarity, initial presentation of flat plaque (skin hardening) and the slow-growing nature of DFSP, it may be months to years without a protuberance (bump). The dissonance between the name of the neoplasm and its clinical presentations may cause a majority of patients to experience a diagnostic delay. A 2019 research study found out of 214 patients a range between less than a year to 42 years before diagnosis (median, four years) from patients noticing a symptom to diagnosis.[24]
Currently, a majority of patients (53%) receive a misdiagnosis by health care providers. The most frequent prebiopsy clinical suspicion included cyst (101 [47.2%]), lipoma (30 [14.0%]), and scar (17 [7.9%]).[24]
It has been suggested an alternative term for DFSP should be dermatofibrosarcoma, often protuberant.[24]
Pregnancy
[edit]It is suggested that DFSPs may enlarge more rapidly during pregnancy. Immunohistochemical stains for CD34, S-100 protein, factor XIIIa, and estrogen and progesterone receptors were performed on biopsy specimens. The tumors showed the expression of the progesterone receptor. As with many other stromal neoplasms, DFSPs appear to express low levels of hormone receptors, which may be one factor that accounts for their accelerated growth during pregnancy.[25]
Treatment
[edit]
Treatment is primarily surgical, with chemotherapy and radiation therapy used if clear resection margins are not acquired.[26]
Surgical treatment
[edit]The type of surgical treatment chosen is dependent on the location of the DFSP occurrence and possible size.
Mohs surgery
[edit]Mohs micrographic surgery (MMS) has a high cure rate and lowers the recurrence reduction of DFSP[27] if negative resection margins are achieved.
Wide local excision
[edit]Wide local excision (WLE) was the gold standard for treating DFSP but is currently under reevaluation. Presently in the United States, WLE may be suggested after the recurrence of MMS. Larger resection margins are suggested for WLE than MMS. Recurrence rate with WLE is about 8.5% with a lower recurrence rate related to wider excision.[28]
Resection margin
[edit]DFSP characteristic features are its capacity to invade surrounding tissues, to a considerable distance from the central focus of the tumor in a "tentacle-like" fashion. This fact, coupled with diagnostic delay, may lead to inadequate initial resection. Inadequate initial treatment results in larger, deeper recurrent lesions, but these can be managed by appropriate wide excision.[29]
Radiation therapy
[edit]DFSP is a radioresponsive tumor; radiation therapy (RT) is not used as the first choice for treatment. Conservative resection through MMS or WLE is attempted first. If clear margins are not achieved RT, or chemotherapy is recommended.[30]
Chemotherapy
[edit]DFSP was previously regarded and nonresponsive to standard chemotherapy.[31] In 2006 the US FDA approved (imatinib mesylate) for the treatment of DFSP.[32] As is true for all medicinal drugs with name ending in "ib," imatinib is a small molecular pathway inhibitor; imatinib inhibits tyrosine kinase. It may be able to induce tumor regression in patients with recurrent DFSP, unresectable DFSP, or metastatic DFSP.[33] There is clinical evidence that imatinib, which inhibits PDGF-receptors, may be effective for tumors positive for the t(17;22) translocation. It is suggested that imatinib may be a treatment for challenging, locally advanced disease and in the rare metastatic cases. It was approved for use by adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans (DFSP).[34]
Metastatic disease
[edit]Distant hematogenous metastases are extremely rare.[35] Metastases to regional lymph nodes are rarer and are most likely in patients who have had multiple local recurrences after inadequate surgical resection.[36] Repeatedly recurring tumors have an increased risk for transformation into a more malignant form (DFSP-FS). The lungs are most frequently affected, but metastases to the brain,[37] bone,[38] and other soft tissues are reported.
Studies
[edit]DFSP is not extensively studied due to its rarity and low mortality. The majority of studies are small size case studies or meta-analysis.
The most extensive research study to date was Perspectives of Patients With Dermatofibrosarcoma Protuberans on Diagnostic Delays, Surgical Outcomes, and Nonprotuberance.[24] The lead researcher, Jerad Gardner, spoke at a TED Talk in February 2020 on the topic.[39]
History
[edit]R. W. Taylor, in 1890,[40] first identified DFSP as a keloid sarcoma. Later in 1924, Ferdinand-Jean Darier and Ferrand identified it as a progressive recurrent dermatofibroma. In 1925, E. Hoffmann[41] coined the term dermatofibrosarcoma protuberans. Bednar tumor was first described by Bednar in 1957.[42][43]
ICD coding
[edit]The following are the ICD-10 medical codes:
- ICD-0: 8832/3[44] – dermatofibrosarcoma protuberans, NOS
- ICD-0: 8833/3[44] – pigmented dermatofibrosarcoma protuberans
- ICD-0: 8834/1[44] – giant cell fibroblastoma
- Fibrosarcomatous dermatofibrosarcoma protuberans: no distinct coding identified
Additional images
[edit]-
 Subcutaneous tissue infiltration (i.e. "honeycomb" growth pattern)
Subcutaneous tissue infiltration (i.e. "honeycomb" growth pattern) -
 Monotonous, plexiform structure of tumour
Monotonous, plexiform structure of tumour -
 DFSP formed both by fibroblastic and histiocytic elements
DFSP formed both by fibroblastic and histiocytic elements -
 Hemosiderin deposits beneath the tumour
Hemosiderin deposits beneath the tumour -
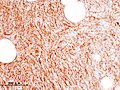 Immunostain positive for CD34
Immunostain positive for CD34




_CD34.JPG)
See also
[edit]References
[edit]- ^ "Dermatofibrosarcoma Protuberans Synonyms & Antonyms". Synonyms.com.
- ^ a b "NCI Dictionary of Cancer Terms". National Cancer Institute. 2011-02-02. Retrieved 2020-06-09.
- ^ "Dermatofibrosarcoma protuberans - Overview - Mayo Clinic". www.mayoclinic.org. Retrieved 2020-06-09.
- ^ Rouhani, Panta; Fletcher, Christopher D. M.; Devesa, Susan S.; Toro, Jorge R. (2008-08-01). "Cutaneous soft tissue sarcoma incidence patterns in the U.S. : an analysis of 12,114 cases". Cancer. 113 (3): 616–627. doi:10.1002/cncr.23571. ISSN 0008-543X. PMID 18618615. S2CID 23746544.
- ^ a b Kreicher, Kathryn L.; Kurlander, David E.; Gittleman, Haley R.; Barnholtz-Sloan, Jill S.; Bordeaux, Jeremy S. (January 2016). "Incidence and Survival of Primary Dermatofibrosarcoma Protuberans in the United States". Dermatologic Surgery. 42 (Suppl 1): S24–31. doi:10.1097/DSS.0000000000000300. ISSN 1524-4725. PMID 26730971. S2CID 12966671.
- ^ Kransdorf, M. J. (January 1995). "Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location". American Journal of Roentgenology. 164 (1): 129–134. doi:10.2214/ajr.164.1.7998525. ISSN 0361-803X. PMID 7998525.
- ^ "Dermatofibrosarcoma protuberans".
- ^ "America's Children and the Environment: Metadata - Surveillance, Epidemiology, and End Results (SEER) Program". United States Environmental Protection Agency. 10 June 2015.
- ^ Sbaraglia M, Bellan E, Dei Tos AP (April 2021). "The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives". Pathologica. 113 (2): 70–84. doi:10.32074/1591-951X-213. PMC 8167394. PMID 33179614.
- ^ Liszewski, Walter; Blanchette, Derek; Cunningham, Ashley M.; Miller, Daniel D. (2016-11-01). "Epidemiology of Bednar tumors in the United States". Journal of the American Academy of Dermatology. 75 (5): 1064–1066. doi:10.1016/j.jaad.2016.06.018. ISSN 0190-9622. PMID 27745635.
- ^ Kaul, Rashmi; Kaur, Navjot; Dogra, Sunder S; Chander, Bal (2015). "Variant of Dermatofibrosarcoma Protuberans: Bednar Tumor". Indian Journal of Dermatology. 60 (1): 107. doi:10.4103/0019-5154.147885. ISSN 0019-5154. PMC 4318047. PMID 25657441.
- ^ Reimann, Julie D. R.; Fletcher, Christopher D. M. (September 2007). "Myxoid dermatofibrosarcoma protuberans: a rare variant analyzed in a series of 23 cases". The American Journal of Surgical Pathology. 31 (9): 1371–1377. doi:10.1097/PAS.0b013e31802ff7e7. ISSN 0147-5185. PMID 17721193. S2CID 1268848.
- ^ a b c d e "Rare Variants of Dermatofibrosarcoma Protuberans (DFSP)". News-Medical.net. 2017-10-24. Retrieved 2020-06-09.
- ^ Buteau, Anna H.; Keeling, Brett H.; Diaz, Lucia Z.; Larralade, Margarita; Luna, Paula; Krishnan, Chandra; Levy, Moise L. (2018-01-16). "Dermatofibrosarcoma protuberans in pediatric patients: A diagnostic and management challenge". JAAD Case Reports. 4 (2): 155–158. doi:10.1016/j.jdcr.2017.09.022. ISSN 2352-5126. PMC 5789519. PMID 29387771.
- ^ "Giant cell fibroblastoma pathology | DermNet NZ". dermnetnz.org. Retrieved 2020-06-09.
- ^ Bakry, Ola; Attia, Abdalla (2012-03-27). "Atrophic dermatofibrosarcoma protuberans". Journal of Dermatological Case Reports. 6 (1): 14–17. doi:10.3315/jdcr.2012.1089. ISSN 1898-7249. PMC 3322104. PMID 22514584.
- ^ Abbott, Jared J.; Oliveira, Andre M.; Nascimento, Antonio G. (April 2006). "The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans". The American Journal of Surgical Pathology. 30 (4): 436–443. doi:10.1097/00000478-200604000-00002. ISSN 0147-5185. PMID 16625088. S2CID 23911032.
- ^ Bowne, W. B.; Antonescu, C. R.; Leung, D. H.; Katz, S. C.; Hawkins, W. G.; Woodruff, J. M.; Brennan, M. F.; Lewis, J. J. (2000-06-15). "Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution". Cancer. 88 (12): 2711–2720. doi:10.1002/1097-0142(20000615)88:12<2711::AID-CNCR9>3.0.CO;2-M. ISSN 0008-543X. PMID 10870053.
- ^ Sirvent N, Maire G, Pedeutour F (May 2003). "Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment". Genes Chromosomes Cancer. 37 (1): 1–19. doi:10.1002/gcc.10202. PMID 12661001. S2CID 36985697.
- ^ Patel KU, Szabo SS, Hernandez VS, et al. (February 2008). "Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays". Hum. Pathol. 39 (2): 184–93. doi:10.1016/j.humpath.2007.06.009. PMID 17950782.
- ^ "What is Dermatofibrosarcoma Protuberans?".
- ^ Serra-Guillén, Carlos; Sanmartín, Onofre; Llombart, Beatriz; Nagore, Eduardo; Deltoro, Carlos; Martín, Isabel; Borella-Estrada, Rafael; Requena, Celia; Martorell-Calatayud, Antonio; Cervera, Jose; Guillén, Carlos (November 2011). "Correlation between preoperative magnetic resonance imaging and surgical margins with modified Mohs for dermatofibrosarcoma protuberans". Dermatologic Surgery. 37 (11): 1638–1645. doi:10.1111/j.1524-4725.2011.02077.x. ISSN 1524-4725. PMID 21679274. S2CID 23878054.
- ^ Kransdorf, M. J.; Meis-Kindblom, J. M. (August 1994). "Dermatofibrosarcoma protuberans: radiologic appearance". American Journal of Roentgenology. 163 (2): 391–394. doi:10.2214/ajr.163.2.8037038. ISSN 0361-803X. PMID 8037038.
- ^ a b c d David, Marjorie Parker; Funderburg, Ashley; Selig, James P.; Brown, Rebecca; Caliskan, Pip M.; Cove, Lee; Dicker, Gayle; Hoffman, Lori; Horne, Tammi; Gardner, Jerad M. (2019-08-30). "Perspectives of Patients With Dermatofibrosarcoma Protuberans on Diagnostic Delays, Surgical Outcomes, and Nonprotuberance". JAMA Network Open. 2 (8): e1910413. doi:10.1001/jamanetworkopen.2019.10413. ISSN 2574-3805. PMC 6724159. PMID 31469398.
- ^ Parlette, L. E.; Smith, C. K.; Germain, L. M.; Rolfe, C. A.; Skelton, H. (November 1999). "Accelerated growth of dermatofibrosarcoma protuberans during pregnancy". Journal of the American Academy of Dermatology. 41 (5 Pt 1): 778–783. doi:10.1016/s0190-9622(99)70023-x. ISSN 0190-9622. PMID 10534646.
- ^ Allen A, Ahn C, Sangüeza OP (October 2019). "Dermatofibrosarcoma Protuberans". Dermatol Clin. 37 (4): 483–488. doi:10.1016/j.det.2019.05.006. PMID 31466588. S2CID 201672710.
- ^ Malan, Malumani; Xuejingzi, Wu; Quan, Song Ji (2019-08-13). "The efficacy of Mohs micrographic surgery over the traditional wide local excision surgery in the cure of dermatofibrosarcoma protuberans". The Pan African Medical Journal. 33: 297. doi:10.11604/pamj.2019.33.297.17692. ISSN 1937-8688. PMC 6815477. PMID 31692830.
- ^ Kim, Byung Jun; Kim, Hyeonwoo; Jin, Ung Sik; Minn, Kyung Won; Chang, Hak (2015). "Wide Local Excision for Dermatofibrosarcoma Protuberans: A Single-Center Series of 90 Patients". BioMed Research International. 2015: 642549. doi:10.1155/2015/642549. ISSN 2314-6133. PMC 4673335. PMID 26688814.
- ^ Khatri, Vijay P.; Galante, Joseph M.; Bold, Richard J.; Schneider, Philip D.; Ramsamooj, Rajendra; Goodnight, James E. (November 2003). "Dermatofibrosarcoma protuberans: reappraisal of wide local excision and impact of inadequate initial treatment". Annals of Surgical Oncology. 10 (9): 1118–1122. doi:10.1245/aso.2003.03.581. ISSN 1068-9265. PMID 14597453. S2CID 41310745.
- ^ Suit, H.; Spiro, I.; Mankin, H. J.; Efird, J.; Rosenberg, A. E. (August 1996). "Radiation in management of patients with dermatofibrosarcoma protuberans". Journal of Clinical Oncology. 14 (8): 2365–2369. doi:10.1200/JCO.1996.14.8.2365. ISSN 0732-183X. PMID 8708729.
- ^ Noujaim, Jonathan; Thway, Khin; Fisher, Cyril; Jones, Robin L. (December 2015). "Dermatofibrosarcoma protuberans: from translocation to targeted therapy". Cancer Biology & Medicine. 12 (4): 375–384. doi:10.7497/j.issn.2095-3941.2015.0067 (inactive 2 December 2024). ISSN 2095-3941. PMC 4706526. PMID 26779374.
{{cite journal}}: CS1 maint: DOI inactive as of December 2024 (link) - ^ "Gleevec Gains Simultaneous FDA Approval for Five Rare, Life-Threatening Disorders". Cancer Network. Oncology NEWS International Vol 15 No 11. 15 (11). 2006-11-01. Archived from the original on 2020-06-10. Retrieved 2020-06-10.
- ^ Rastogi, Sameer; Dhamija, Ekta; Barwad, Adarsh; Aggarwal, Aditi; Sharma, Atul; Panday, Rambha (December 2018). "Advanced Dermatofibrosarcoma Protuberans Treatment With Imatinib: Experience From a Dedicated Sarcoma Medical Oncology Clinic in India". Journal of Global Oncology. 4 (4): 1–7. doi:10.1200/JGO.18.00007. PMC 6223511. PMID 30085879.
- ^ "Lupin in alliance with Natco receives FDA approval for Imatinib Mesylate Tablets". Lupin. 5 March 2019. Retrieved 2020-06-10.
- ^ Mavili, M. E.; Gursu, K. G.; Gokoz, A. (April 1994). "Dermatofibrosarcoma with lymph node involvement". Annals of Plastic Surgery. 32 (4): 438–440. doi:10.1097/00000637-199404000-00022. ISSN 0148-7043. PMID 8210168.
- ^ Rutgers, E. J.; Kroon, B. B.; Albus-Lutter, C. E.; Gortzak, E. (June 1992). "Dermatofibrosarcoma protuberans: treatment and prognosis". European Journal of Surgical Oncology: The Journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 18 (3): 241–248. ISSN 0748-7983. PMID 1607035.
- ^ Mahajan, B. B.; Sumir, Kumar; Singla, Monika (July 2015). "Metastatic dermatofibrosarcoma protuberans: A rare case report from North India". Journal of Cancer Research and Therapeutics. 11 (3): 670. doi:10.4103/0973-1482.146099. ISSN 1998-4138. PMID 26458720.
- ^ Garg, Mandeep Kumar; Yadav, Mukesh Kumar; Gupta, Suruchi; Kumar, Narender; Khandelwal, Niranjan (2009-09-29). "Dermatofibrosarcoma protuberans with contiguous infiltration of the underlying bone". Cancer Imaging. 9 (1): 63–66. doi:10.1102/1470-7330.2009.0011 (inactive 1 November 2024). ISSN 1740-5025. PMC 2792083. PMID 19933019.
{{cite journal}}: CS1 maint: DOI inactive as of November 2024 (link) - ^ Gardner, Jerad (2020-06-08). "Facebook and Rare Cancer Changed My Life". TED Conferences. Retrieved 2023-09-18.
- ^ Shen, Kuang-Hsuan; Leu, Yi-Shing (2017-09-01). "Dermatofibrosarcoma protuberans of the cheek". Journal of Cancer Research and Practice. 4 (3): 119–121. doi:10.1016/j.jcrpr.2017.03.001. ISSN 2311-3006.
- ^ Hoffmann, Erich (1925). "I. Über das knollentreibende Fibrosarkom der Haut (Dermatofibrosarkoma protuberans)". Dermatology. 43 (1–2): 1–28. doi:10.1159/000250699. ISSN 1018-8665.
- ^ "Bednar tumour: an infrequent diagnosis | British Journal of Medical Practitioners". www.bjmp.org. Retrieved 2020-06-09.
- ^ Bednar, B. (March 1957). "Storiform neurofibromas of the skin, pigmented and nonpigmented". Cancer. 10 (2): 368–376. doi:10.1002/1097-0142(195703/04)10:2<368::aid-cncr2820100218>3.0.co;2-3. ISSN 0008-543X. PMID 13426994. S2CID 42490204.
- ^ a b c "Dermatofibrosarcoma protuberans (DFSP)". www.pathologyoutlines.com. Retrieved 2020-06-09.
External links
[edit]- Dermatofibrosarcoma protuberans in NIH Genetic and Rare Diseases Information Center