
An enamine is an unsaturated compound derived by the condensation of an aldehyde or ketone with a secondary amine.[1][2] Enamines are versatile intermediates.[3][4]
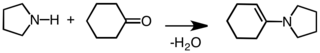
Condensation to give an enamine.[5]

The word "enamine" is derived from the affix en-, used as the suffix of alkene, and the root amine. This can be compared with enol, which is a functional group containing both alkene (en-) and alcohol (-ol). Enamines are considered to be nitrogen analogs of enols.[6]
If one or both of the nitrogen substituents is a hydrogen atom it is the tautomeric form of an imine. This usually will rearrange to the imine; however there are several exceptions (such as aniline). The enamine-imine tautomerism may be considered analogous to the keto-enol tautomerism. In both cases, a hydrogen atom switches its location between the heteroatom (oxygen or nitrogen) and the second carbon atom.
Enamines are both good nucleophiles and good bases. Their behavior as carbon-based nucleophiles is explained with reference to the following resonance structures.

Formation
[edit]Enamines are labile and therefore chemically useful moieties which can be easily produced from commercially available starting reagents. A common route for enamine production is via an acid-catalyzed nucleophilic reaction of ketone[7] or aldehyde[8] species containing an α-hydrogen with secondary amines. Acid catalysis is not always required, if the pKaH of the reacting amine is sufficiently high (for example, pyrrolidine, which has a pKaH of 11.26). If the pKaH of the reacting amine is low, however, then acid catalysis is required through both the addition and the dehydration steps[9] (common dehydrating agents include MgSO4 and Na2SO4).[10] Primary amines are usually not used for enamine synthesis due to the preferential formation of the more thermodynamically stable imine species.[11] Methyl ketone self-condensation is a side-reaction which can be avoided through the addition of TiCl4[12] into the reaction mixture (to act as a water scavenger).[13][14] An example of an aldehyde reacting with a secondary amine to form an enamine via a carbinolamine intermediate is shown below:

Reactions
[edit]Alkylation
[edit]Even though enamines are more nucleophilic than their enol counterparts, they can still react selectively, rendering them useful for alkylation reactions. The enamine nucleophile can attack haloalkanes to form the alkylated iminium salt intermediate which then hydrolyzes to regenerate a ketone (a starting material in enamine synthesis). This reaction was pioneered by Gilbert Stork, and is sometimes referred to by the name of its inventor (the Stork enamine alkylation). Analogously, this reaction can be used as an effective means of acylation. A variety of alkylating and acylating agents including benzylic, allylic halides can be used in this reaction.[15]

Acylation
[edit]In a reaction much similar to the enamine alkylation, enamines can be acylated to form a final dicarbonyl product. The enamine starting material undergoes a nucleophilic addition to acyl halides forming the iminium salt intermediate which can hydrolyze in the presence of acid.[16]

Metalloenamines
[edit]Strong bases such as LiNR2 can be used to deprotonate imines and form metalloenamines. Metalloenamines can prove synthetically useful due to their nucleophilicity (they are more nucleophilic than enolates). Thus they are better able to react with weaker electrophiles (for example, they can be used to open epoxides.[17]) Most prominently, these reactions have allowed for asymmetric alkylations of ketones through transformation to chiral intermediate metalloenamines.[18]
Halogenation
[edit]β-halo immonium compounds can be synthesized through the halogenation reaction of enamines with halides in diethyl ether solvent. Hydrolysis will result in the formation of α-halo ketones.[19] Chlorination, bromination, and even iodination have been shown to be possible. The general reaction is shown below:

Oxidative coupling
[edit]Enamines can be efficiently cross-coupled with enol silanes through treatment with ceric ammonium nitrate. These reactions were reported by the Narasaka group in 1975, providing a route to stable enamines as well as one instance of a 1,4-diketone (derived from a morpholine amine reagent).[20] Later, these results were exploited by the MacMillan group with the development of an organocatalyst which used the Narasaka substrates to produce 1,4 dicarbonyls enantioselectively, with good yields.[21] Oxidative dimerization of aldehydes in the presence of amines proceeds through the formation of an enamine followed by a final pyrrole formation.[22] This method for symmetric pyrrole synthesis was developed in 2010 by the Jia group, as a valuable new pathway for the synthesis of pyrrole-containing natural products.[23]
Annulation
[edit]Enamines chemistry has been implemented for the purposes of producing a one-pot enantioselective version of the Robinson annulation. The Robinson annulation, published by Robert Robinson in 1935, is a base-catalyzed reaction that combines a ketone and a methyl vinyl ketone (commonly abbreviated to MVK) to form a cyclohexenone fused ring system. This reaction may be catalyzed by proline to proceed through chiral enamine intermediates which allow for good stereoselectivity.[24] This is important, in particular in the field of natural product synthesis, for example, for the synthesis of the Wieland-Miescher ketone – a vital building block for more complex biologically active molecules.[25][26]
Reactivity
[edit]Enamines act as nucleophiles that require less acid/base activation for reactivity than their enolate counterparts. They have also been shown to offer a greater selectivity with less side reactions. There is a gradient of reactivity among different enamine types, with a greater reactivity offered by ketone enamines than their aldehyde counterparts.[27] Cyclic ketone enamines follow a reactivity trend where the five membered ring is the most reactive due to its maximally planar conformation at the nitrogen, following the trend 5>8>6>7 (the seven membered ring being the least reactive). This trend has been attributed to the amount of p-character on the nitrogen lone pair orbital - the higher p character corresponding to a greater nucleophilicity because the p-orbital would allow for donation into the alkene π- orbital. Analogously, if the N lone pair participates in stereoelectronic interactions on the amine moiety, the lone pair will pop out of the plane (will pyramidalize) and compromise donation into the adjacent π C-C bond.[28][29]

There are many ways to modulate enamine reactivity in addition to altering the steric/electronics at the nitrogen center including changing temperature, solvent, amounts of other reagents, and type of electrophile. Tuning these parameters allows for the preferential formation of E/Z enamines and also affects the formation of the more/less substituted enamine from the ketone starting material.[30]
See also
[edit]- Enders SAMP/RAMP hydrazone-alkylation reaction
- Hajos–Parrish–Eder–Sauer–Wiechert reaction
- Michael Addition
- Nenitzescu indole synthesis
- Organocatalysis
- Robinson annulation
- Thorpe reaction
- Fluoxymesterone
References
[edit]- ^ Clayden, Jonathan (2001). Organic chemistry. Oxford, Oxfordshire: Oxford University Press. ISBN 978-0-19-850346-0.
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Enamines: Synthesis: Structure, and Reactions, Second Edition, Gilbert Cook (Editor). 1988, Marcel Dekker, NY. ISBN 0-8247-7764-6
- ^ R. B. Woodward, I. J. Pachter, and M. L. Scheinbaum (1974). "2,2- (Trimethylenedithio)cyclohexanone". Organic Syntheses. 54: 39
{{cite journal}}: CS1 maint: multiple names: authors list (link); Collected Volumes, vol. 5, p. 1014. - ^ R. D. Burpitt and J. G. Thweatt (1968). "Cyclodecanone". Organic Syntheses. 48: 56; Collected Volumes, vol. 5, p. 277.
- ^ Imines and Enamines | PharmaXChange.info
- ^ Stork, Gilbert.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. (1963). "The Enamine Alkylation and Acylation of Carbonyl Compounds". Journal of the American Chemical Society. 85 (2): 207–222. doi:10.1021/ja00885a021. ISSN 0002-7863.
- ^ Mannich, C.; Davidsen, H. (1936). "Über einfache Enamine mit tertiär gebundenem Stickstoff" [On simple enamines with triple-bonded nitrogen]. Berichte der Deutschen Chemischen Gesellschaft (A and B Series) (in German). 69 (9): 2106–2112. doi:10.1002/cber.19360690921. ISSN 0365-9488.
- ^ Capon, Brian; Wu, Zhen Ping (April 1990). "Comparison of the tautomerization and hydrolysis of some secondary and tertiary enamines". The Journal of Organic Chemistry. 55 (8): 2317–2324. doi:10.1021/jo00295a017.
- ^ Lockner, James. "Stoichiometric Enamine Chemistry" (PDF). Baran Group, The Scripps Research Institute. Retrieved 26 November 2014.
- ^ Farmer, Steven (2013-10-16). "Enamine Reactions". UC Davis Chem Wiki.
- ^ Carlson, R; Nilsson, A (1984). "Improved Titanium Tetrachloride Procedure for Enamine Synthesis". Acta Chemica Scandinavica. 38B: 49–53. doi:10.3891/acta.chem.scand.38b-0049.
- ^ Lockner, James. "Stoichiometric Enamine Chemistry" (PDF). Baran Group, The Scripps Research Institute. Retrieved 26 November 2014.
- ^ White, William Andrew; Weingarten, Harold (January 1967). "A versatile new enamine synthesis". The Journal of Organic Chemistry. 32 (1): 213–214. doi:10.1021/jo01277a052.
- ^ Wade, L.G. (1999). Organic Chemistry. Saddle River, NJ: Prentice Hall. pp. 1019. ISBN 9780139227417.
- ^ Farmer, Steven (2013-10-16). "Enamine Reactions". UC Davis Chem Wiki.
- ^ Evans, D. "Enolates and Metalloenamines II" (PDF). Retrieved 10 December 2014.[permanent dead link]
- ^ Meyers, A. I.; Williams, Donald R. (August 1978). "Asymmetric alkylation of acyclic ketones via chiral metallo enamines. Effect of kinetic vs. thermodynamic metalations". The Journal of Organic Chemistry. 43 (16): 3245–3247. doi:10.1021/jo00410a034.
- ^ Seufert, Walter; Eiffenberger, Franz (1979). "Zur Halogenierung von Enaminen — Darstellung von β-Halogen-iminium-halogeniden". Chemische Berichte. 112 (5): 1670–1676. doi:10.1002/cber.19791120517.
- ^ Ito, Y; Konoike, T; Saegusa, T (1975). "Synthesis of 1,4-diketones by the reaction of silyl enol ether with silver oxide. Regiospecific formation of silver(I) enolate intermediates". Journal of the American Chemical Society. 97 (3): 649–651. doi:10.1021/ja00836a034.
- ^ Jang, HY; Hong, JB; MacMillan, DWC (2007). "Enantioselective organocatalytic singly occupied molecular orbital activation: the enantioselective alpha-enolation of aldehydes" (PDF). J. Am. Chem. Soc. 129 (22): 7004–7005. doi:10.1021/ja0719428. PMID 17497866.
- ^ Li, Q; Fan, A; Lu, Z; Cui, Y; Lin, W; Jia, Y (2010). "One-pot AgOAc-mediated synthesis of polysubstituted pyrroles from primary amines and aldehydes: application to the total synthesis of purpurone". Organic Letters. 12 (18): 4066–4069. doi:10.1021/ol101644g. PMID 20734981.
- ^ Guo, Fenghai; Clift, Michael D.; Thomson, Regan J. (September 2012). "Oxidative Coupling of Enolates, Enol Silanes, and Enamines: Methods and Natural Product Synthesis". European Journal of Organic Chemistry. 2012 (26): 4881–4896. doi:10.1002/ejoc.201200665. PMC 3586739. PMID 23471479.
- ^ List, Benjamin (2002). "Proline-catalyzed asymmetric reactions". Tetrahedron. 58 (28): 5573–5590. doi:10.1016/s0040-4020(02)00516-1.
- ^ Bui, Tommy; Barbas (2000). "A proline-catalyzed asymmetric Robinson Annulation". Tetrahedron Letters. 41 (36): 6951–6954. doi:10.1016/s0040-4039(00)01180-1.
- ^ Wiener, Jake. "Enantioselective Organic Catalysis:Non-MacMillan Approaches" (PDF). Archived from the original (PDF) on 26 October 2017. Retrieved 29 November 2014.
- ^ Hickmott, Peter (May 1982). "Enamines: Recent advances in synthetic, spectroscopic, mechanistic, and stereochemical aspects—II". Tetrahedron. 38 (23): 3363–3446. doi:10.1016/0040-4020(82)85027-8.
- ^ Mayr, H. (2003). "Structure-Nucleophilicity Relationships for Enamines". Chem. Eur. J. 9 (10): 2209–18. doi:10.1002/chem.200204666. PMID 12772295.
- ^ Hickmott, Peter (May 1982). "Enamines: Recent advances in synthetic, spectroscopic, mechanistic, and stereochemical aspects—II". Tetrahedron. 38 (23): 3363–3446. doi:10.1016/0040-4020(82)85027-8.
- ^ Lockner, James. "Stoichiometric Enamine Chemistry" (PDF). Baran Group, The Scripps Research Institute. Retrieved 26 November 2014.
| Authority control databases: National |
|---|